
The new website: Yakhini Research Group (yakhini-research-group.org)
The recent progress in high throughput measurement technologies for molecular biology, such as high throughput sequencing, microarrays, multi-dimensional proteomics, glycomics and metabolomics is driving modern biology to heavily overlap data and information sciences. Computational biology and bioinformatics are research fields that focus on developing efficient and effective data analysis and design algorithmics to address data science challenges presented by the fast progress of modern molecular biology and on applying these to obtain novel biological insights.
Design and data analysis methods are playing a central role in enabling molecular based personalized medicine. They also play a role in the development of synthetic biology, driving our ability to efficiently design and utilize molecular devices.
Mining healthcare related data in various levels and from different organizations is fast emerging as a tool for improving patient care and human wellbeing.
The algorithmic and statistical aspects of these developing fields of science present diverse, deep and fascinating challenges.
Our group develops statistical and algorithmic methods for analyzing medical and molecular measurement data. We also develop optimization and design algorithmics for measurement systems or for assays and reagents to increase efficiency and effectiveness. For example: we developed an efficient scheme for storing data using DNA and a measurement/analysis approach for quantifying CRISPR genome editing activity. Both of these represent on-going research directions in the group.
We often implement the methods into software tools that serve a broad scientific community.
In collaboration with the FACT center at IDC we are working on cryptographic techniques to enable machine learning and statistical inference in a multiparty computation framework.
We also apply data science, machine learning and statistical techniques to data from other domains.
More detailed descriptions of some of our projects can be found below.
- Synthetic biology and genome editing.
We are interested in the design of reagents, application development and data analysis related to synthetic biology and genetic engineering in general.- CRISPR off target activity assessment.
Applying a Bayes classification approach and other statistical methods we developed software, called CRISPECTOR, for analyzing genome editing experiments and evaluating off target activi ty. Notably, CRISPECTOR supports the identification of indel activity as well as of translocations.
ty. Notably, CRISPECTOR supports the identification of indel activity as well as of translocations. - DNA based data storage
DNA based storage systems represent an appealing potential solution for the surging demand for digital information storage. We develop novel methodologies to utilize the properties of synthetic DNA in order to increase the capacity and reduce the costs of such systems. In our recent publication in Nature Biotechnology we introduced the use of composite DNA letters to increase the logical density of DNA storage above the strict, single molecule, theoretical limit of 2 bits per synthesis cycle.
[Read More] - Oligonucleotide Library (OL) based assays
Oligonucleotide libraries consist of a high multiplicity (tens of thousands) of short synthetic DNA molecules that are designed to perform a certain task or investigate a certain phenomenon or a set of them.
[Read More] - QC and statistics for OLs
In collaboration with Eitan Yaakobi’s group we are developing methods and tools for analyzing synthetic oligo libraries and for inferring and reporting library characteristics. This work was accepted to be presented at the NVMW 2020 conference. - Flux balance analysis (FBA)
Metabolic models of organisms can help design and optimize expression and production systems. One of our current active projects addresses the combination of several organisms in a fermentation process designed to optimize ethanol production (Vitkin et al Technology 2015, Jiang et al Sci Rep 2016). We also used high throughput fitness measurement results to infer genes coding to orphan E coli model reactions (Vitkin, Solomon et al, BMC Bioinformatics 2018).
- CRISPR off target activity assessment.
- Statistics in ranked lists
Methods and tools to enable inference in noisy datasets. Related projects include:- Minimum Hypergeometric Statistics (mHG)
Consider a ranked list of elements and a binary labeling associated with all elements in the list. The mHG statistic measures the density of 1s at the top of the resulting binary vector. Our work included the full characterization of the distribution of mHG assuming a uniform null model (Eden et al, PLoS CB 2007), the development of specialized tools that use mHG (see below) and the development of variants, generalizations and extensions (Leibovich et al, NAR 2012; Steinfeld et al, NAR 2013) - GOrilla: a software tool for identifying and visualizing enriched GO terms in ranked lists of genes, based on the mHG statistics. (Eden et al, BMC Bioinformatics, 2009)
-
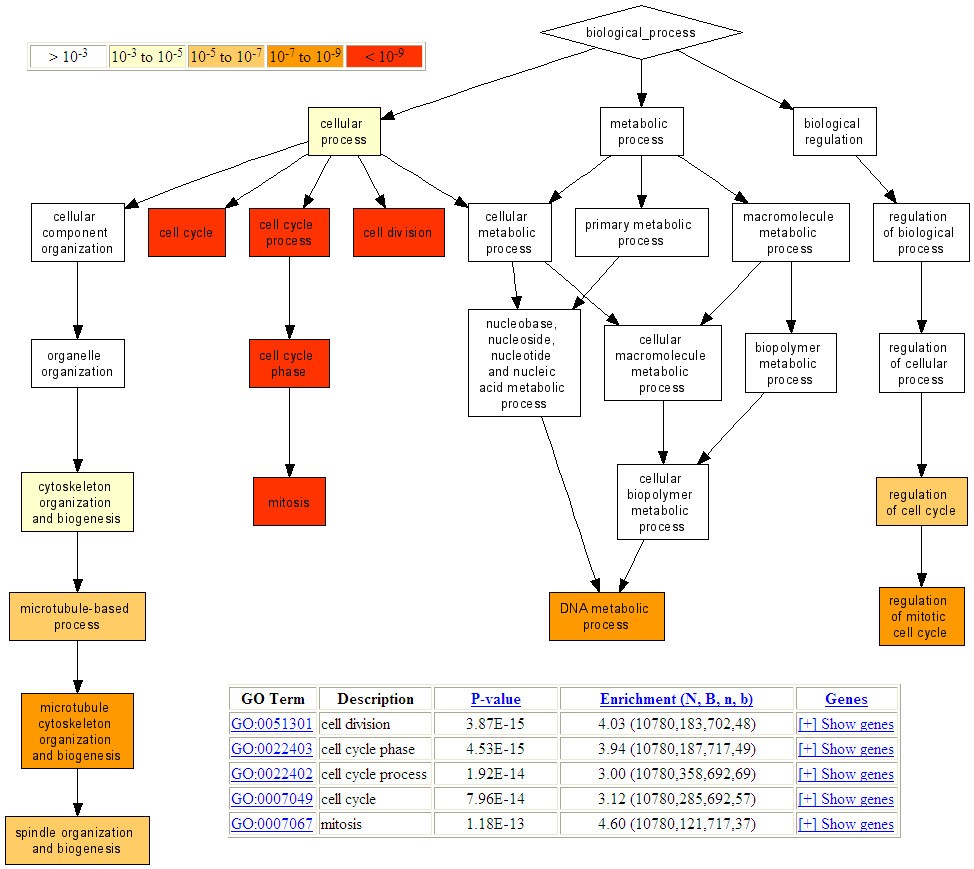 DRIMust: a software tool for identifying enriched sequence motifs in ranked lists of sequences.
DRIMust: a software tool for identifying enriched sequence motifs in ranked lists of sequences. - miTEA and MULSEA: software tools that support the analysis of miRNA targets in ranked lists of genes. (Steinfeld et al, NAR 2013; Cohn-Alperovich et al, Bioinformatics, 2016)
- Minimum Hypergeometric Statistics (mHG)
- Privacy preserving machine learning
In work with Adi Akavia (Haifa Univ), Hayim Shaul (IDC) and Mor Weiss (IDC) we developed a protocol for privacy preserving linear regression (WHAC 2019). We are currently working on extending this approach to enable more privacy preserving machine learning algorithms. - Epigenetics
Design of reagents, application development and data analysis as related to various aspects of molecular regulation in living cells.- DNA methylation
In collaborations with the Cedar Lab at HUJI we investigated DNA methylation and its sequence determinants (Schlesinger et al Nat Gen 2007, Straussman et al Nat SMB 2009). We further investigated the relationship between age related DNA methylation and silencing that is related to cancer (Nejman et al Cancer Res 2014). - Deep learning models for the prediction of the methylation status of individual CpGs
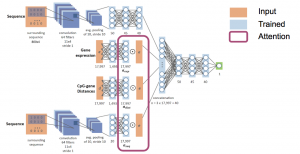 We produced a model that predicts DNA methylation for a given sample in any CpG position based solely on the sample’s gene expression profile and the sequence surrounding the CpG. Our approach offers a novel framework with which to extract valuable insights from gene expression data when combined with sequence information.
We produced a model that predicts DNA methylation for a given sample in any CpG position based solely on the sample’s gene expression profile and the sequence surrounding the CpG. Our approach offers a novel framework with which to extract valuable insights from gene expression data when combined with sequence information. - Time of replication (ToR)
In collaboration with the Simon Lab at HUJI we investigated time of replication and associated properties of genomic regions in mice and men (Farkash-Amar et al Gen Res 2008, Farkash-Amar et al, PLoS One 2012). Arto is a software tool that support analyzing ToR data. - HiC data
We are interested in general embedding techniques (MDS and NMDS) and their application to HiC data. In Ben Elazar et al NAR 2013, Ben Elazar et al Bioinformatics 2016 and Ben-Elazar et al Sci Rep 2019 we report related results, including haplotyping methods as well as statistical enrichment that elucidates the 3D functional organization of unicellular genomes.
- DNA methylation
- Computational biology in cancer and other disease
- Collaborative research in medical science
We develop statistical analysis and algorithms to support, improve and sometimes drive medical science studies, in collaboration with leading molecular medicine groups. - Digital pathology and spatial transcriptomics
In recent work we demonstrated the use of deep learning to infer molecular cartography of the tumor micro-environment from H&E images. We train a weakly-supervised model using bulk molecular and then use the model to infer molecular traits on WSIs. (Levy-Jurgensen et al, Sci Rep 2020)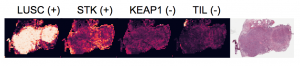
- Data integration and joint analysis of data from several different platforms
Methods and tools to enhance interpretation and inference in rich datasets. In breast cancer we studied, for example, the direct activity of miRNA as driving disease related processes (Enerly, Steinfeld et al PLoS One 2011). - Cross platform normalization for miRNA in breast cancer
As described in Ben-Elazar et al PLoS Comp Bio 2021. - Glycomics
We studied the association of serum glycans to molecular processes in the tumor (Haakensen et al Mol Onc 2016). - Data analysis for single cell RNA-Seq and spatial transcriptomics
Investigating questions related to the interpretation of emerging sequencing technologies. In particular, we developed analysis methods for spatial transcriptomics data. Measures of heterogeneity are described in Levy-Jurgenson et al 2020.
- Collaborative research in medical science
- Metabolomics and computational aspects of environmental science
In collaboration with Alex Golberg’s Lab at TAU we are working on optimizing and investigating various aspects of biorefinery designed and related metabolic measurements. Vitkin et al PLoS One 2020 and Ingle et al BioE Res 2020, for example, address the production of fuel from readily available algae. In Peleg et al Analytica Chim Acta 2019, we investigate the efficient characterization of analyte composition from NIR spectra. - Machine learning and data science: methods and applications
- Interpolation in the latent representation of images
In recent work (Oring et al ICML 2021) we developed an approach to interpolate images in a low dimensional latent space, with minimal loss. - Stealth detection in computer network
- IoT or NOT
Levy et al 2020 demonstrates the use of machine learning on packet traffic to determine whether a newly installed device is IoT or Not.
- Interpolation in the latent representation of images
- Improved assay design and inference
We apply algorithmics and statistical approaches to optimize assay components and to assess performance. Examples include:- CGH probes (Barret et al PNAS 2004, Lipson et al Bioinformatics 2007)
- Optimized IEF-LC/MS (Kifer et al IEEE Bioinformatics 2017).
- SNIRO (Peleg et al Analytica Chim Acta 2019)
- CRISPECTOR.A software tool for analyzing CRISPR performance (Amit et al, Nat Comm 2021).